
Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Lissencephaly otak
Pakar perubatan artikel itu

Ulasan terakhir: 04.07.2025
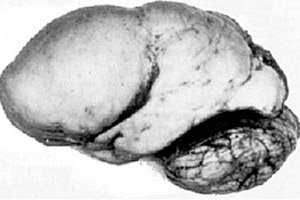
Di antara patologi serebrum organik, anomali kongenital perkembangan otak seperti lissencephaly menonjol, intipatinya terletak pada permukaan hampir licin korteks hemisferanya - dengan bilangan belitan dan alur yang tidak mencukupi. [ 1 ]
Dalam ketiadaan konvolusi sepenuhnya, agyria ditakrifkan, dan kehadiran beberapa belitan rata yang luas dipanggil pachygyria. Kecacatan ini, seperti beberapa ubah bentuk pengurangan otak yang lain, mempunyai kod Q04.3 dalam ICD-10.
Epidemiologi
Menurut statistik mengenai penyakit jarang berlaku, terdapat 1-1.2 kes lissencephaly setiap 100 ribu bayi baru lahir. [ 2 ], [ 3 ]
Menurut beberapa data, sehingga 25-30% kes lissencephaly klasik diperhatikan pada kanak-kanak dengan sindrom Miller-Dieker; mutasi titik dan pemadaman gen LIS1 dan DCX dikesan dalam hampir 85% pesakit. [ 4 ]
Kajian genetik terhadap 17 gen yang dikaitkan dengan lissencephaly menunjukkan bahawa mutasi atau pemadaman LIS1 menyumbang 40% pesakit, dan 23% dikaitkan dengan mutasi DCX, diikuti oleh TUBA1A (5%) dan DYNC1H1 (3%).[ 5 ]
Punca lissencephaly
Semua punca pembentukan korteks serebrum (korteks cerebri) yang diketahui hampir atau sepenuhnya tanpa konvolusi dan alur, yang meningkatkan "kawasan kerja" otak manusia dan memastikan "produktiviti" sistem saraf pusat, dikaitkan dengan gangguan dalam perkembangan perinatalnya. Iaitu, lissencephaly berkembang dalam janin. [ 6 ]
Kegagalan untuk membentuk lapisan korteks serebrum janin dalam lissencephaly adalah hasil daripada penghijrahan abnormal neuron yang membentuknya atau pemberhentian pramatang proses ini.
Proses ini, yang penting untuk histogenesis serebrokortikal, berlaku dalam beberapa peringkat dari minggu ke-7 hingga ke-18 kehamilan. Dan, memandangkan peningkatan kepekaannya terhadap mutasi genetik, serta pelbagai pengaruh fizikal, kimia dan biologi negatif, sebarang penyelewengan dari norma boleh membawa kepada penyetempatan neuron yang tidak betul dengan kemungkinan pembentukan lapisan tebal bahan kelabu korteks tanpa struktur ciri. [ 7 ]
Dalam sesetengah kes, lissencephaly pada kanak-kanak dikaitkan dengan sindrom Miller-Dieker, Walker-Warburg, atau Norman-Roberts.
Baca juga – Kecacatan perkembangan otak
Faktor-faktor risiko
Sebagai tambahan kepada mutasi dalam sesetengah gen, faktor risiko untuk kelahiran kanak-kanak dengan kecacatan yang teruk termasuk kebuluran oksigen (hipoksia) janin; bekalan darah yang tidak mencukupi ke otak (hipoperfusi); kemalangan serebrovaskular akut dalam bentuk strok perinatal; patologi plasenta; jangkitan virus wanita hamil (termasuk TORCH); [ 8 ] masalah dengan metabolisme umum dan fungsi tiroid; merokok, alkohol, psikotropik dan bahan narkotik; penggunaan beberapa ubat; peningkatan tahap sinaran. [ 9 ]
Patogenesis
Tidak semua kes lissencephaly mempunyai patogenesis yang disebabkan oleh keabnormalan kromosom dan mutasi gen. Tetapi beberapa gen diketahui yang mengekod protein yang memainkan peranan penting dalam pergerakan neuroblas dan neuron yang betul di sepanjang sel glia radial - untuk membentuk korteks serebrum. Dan mutasi gen ini membawa kepada patologi ini. [ 10 ]
Khususnya, ini adalah mutasi sporadis (tanpa keturunan) gen LIS1 pada kromosom 17, yang mengawal protein motor sitoplasma microtubules dynein, serta gen DCX pada kromosom X, yang mengkodkan protein doublecortin (lissencephalin-X). [ 11 ]. Dalam kes pertama, pakar mentakrifkan lissencephaly klasik (jenis I), dalam kes kedua - X-linked. [ 12 ]
Apabila gen FLN1, yang mengekodkan phosphoprotein filamin 1, dipadamkan, proses penghijrahan neuron terarah mungkin tidak bermula sama sekali, membawa kepada ketiadaan konvolusi (agyria) sepenuhnya. [ 13 ]
Mutasi telah dikenal pasti dalam gen CDK5, yang mengekod enzim kinase - pemangkin untuk metabolisme intraselular, mengawal kitaran sel dalam neuron CNS dan memastikan penghijrahan normal mereka semasa pembentukan struktur otak pranatal.
Perubahan abnormal dalam gen RELN pada kromosom 7 yang menyebabkan kecacatan gyral kortikal dalam sindrom Norman-Roberts mengakibatkan kekurangan reelin glikoprotein ekstraselular, yang diperlukan untuk mengawal penghijrahan dan kedudukan sel stem neural semasa perkembangan korteks serebri.[ 14 ], [ 15 ], [ 16 ]
Gen ARX mengekod protein homeobox bukan aristalens, faktor transkripsi yang memainkan peranan penting dalam otak depan dan tisu lain.[ 17 ] Kanak-kanak dengan mutasi ARX mempunyai gejala lain, seperti bahagian otak yang hilang (agenesis corpus callosum), alat kelamin yang tidak normal, dan epilepsi yang teruk.[ 18 ],[ 19 ]
Beberapa gen telah dikaitkan dengan lissencephaly. Gen ini termasuk VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A dan CDK5.[ 20 ]
Cytomegalovirus (CMV) telah dikaitkan dengan perkembangan lissencephaly akibat penurunan bekalan darah ke otak janin. Keterukan jangkitan CMV bergantung pada usia kehamilan. Jangkitan awal lebih berkemungkinan menyebabkan lissencephaly kerana penghijrahan neuron berlaku pada awal kehamilan.[ 21 ]
Di samping itu, mekanisme kejadian anomali ini termasuk pemberhentian tidak lengkap atau kemudiannya penghijrahan neuron dari zon generatif periventrikular ke korteks serebrum. Dan dalam kes sedemikian, sama ada lissencephaly atau pachygyria yang tidak lengkap berkembang, di mana beberapa alur dan konvolusi yang luas terbentuk (tetapi kebanyakannya tidak hadir).
Gejala lissencephaly
Tanda-tanda pertama patologi ini (jika tiada sindrom yang disebutkan sebelumnya) mungkin tidak muncul serta-merta selepas kelahiran, tetapi selepas satu setengah hingga dua bulan. Dan selalunya, gejala klinikal lissencephaly berikut diperhatikan:
- hipotonia otot, sering digabungkan dengan lumpuh spastik;
- sawan dan sawan tonik-klonik umum (dalam bentuk opisthotonus);
- terencat akal yang teruk dan terencat pertumbuhan;
- kemerosotan fungsi saraf dan motor.
Masalah menelan menyebabkan kesukaran menyusukan bayi. [ 22 ]
Tahap kecacatan neuromotor yang tinggi sering menunjukkan dirinya sebagai tetraplegia - lumpuh semua anggota badan. Ubah bentuk tangan, jari tangan atau kaki adalah mungkin.
Dalam sindrom Norman-Roberts dengan jenis lissencephaly I, anomali kraniofasial diperhatikan: mikrosefali teruk, cerun dahi rendah dan jambatan hidung lebar yang menonjol, mata lebar (hiperterlorisme), keterbelakangan rahang (micrognathia). [ 23 ]
Sindrom Miller-Dieker juga boleh dicirikan oleh saiz kepala yang luar biasa kecil dengan dahi yang lebar, tinggi dan hidung pendek, lekukan pada pelipis (bitemporal depression), dan set rendah, telinga yang cacat.
Sindrom lissencephaly yang teruk dicirikan oleh microcephaly, pengurangan saiz bola mata (microphthalmia), digabungkan dengan displasia retina, hidrosefalus obstruktif, dan korpus callosum tidak hadir atau hipoplastik.
Komplikasi dan akibatnya
Antara komplikasi anomali ini, pakar menamakan pelanggaran fungsi menelan (disfagia) dan refluks gastroesophageal; epilepsi refraktori (tidak terkawal); jangkitan saluran pernafasan atas yang kerap; radang paru-paru (termasuk aspirasi kronik).
Bayi dengan lissencephaly mungkin mengalami masalah jantung organik kongenital dalam bentuk kecacatan septum atrium atau kecacatan jantung kompleks dengan sianosis (tetralogi Fallot). [ 24 ]
Akibat keterlambatan pertumbuhan selepas bersalin dalam kebanyakan kes mengakibatkan kematian dalam tempoh 24 bulan selepas kelahiran.
Diagnostik lissencephaly
Diagnosis bermula dengan pemeriksaan fizikal kanak-kanak, kajian sejarah perubatan ibu bapa dan sejarah kehamilan dan melahirkan anak.
Semasa kehamilan, ujian DNA janin tanpa sel, amniosentesis atau pensampelan vilus korionik mungkin diperlukan. [ 25 ] Untuk maklumat lanjut, lihat – Diagnosis Pranatal Penyakit Kongenital
Diagnostik instrumental digunakan untuk menggambarkan struktur otak dan menilai fungsinya:
- tomografi otak yang dikira;
- pengimejan resonans magnetik (MRI) otak;
- elektroensefalogram (EEG). [ 26 ]
Semasa kehamilan, lissencephaly pada ultrasound janin selepas 20-21 minggu mungkin disyaki jika tiada alur parieto-occipital dan calcarine dan anomali fisur Sylvian otak.
Diagnosis pembezaan
Diagnostik pembezaan dengan sindrom lain kecacatan serebrum kongenital dijalankan.
Terdapat lebih daripada 20 jenis lissencephaly, kebanyakannya terbahagi kepada 2 kategori utama: lissencephaly klasik (jenis 1) dan lissencephaly batu bulat (jenis 2). Setiap kategori mempunyai manifestasi klinikal yang sama tetapi mutasi genetik yang berbeza.[ 27 ]
Pemeriksaan otak dalam lissencephaly jenis I menunjukkan korteks serebrum dengan empat lapisan dan bukannya enam yang dilihat pada pesakit normal, manakala dalam lissencephaly jenis 2 korteks serebrum tidak teratur dan kelihatan berketul atau bernodular kerana anjakan lengkap korteks serebrum oleh gugusan neuron kortikal yang dipisahkan oleh tisu gliomesenchymal. Pesakit juga mempunyai keabnormalan otot dan mata.
- Lissencephaly klasik (jenis 1):
- LIS1: Lissencephaly terpencil dan sindrom Miller-Dieker (lissencephaly dikaitkan dengan dismorfisme muka). [ 28 ]
- LISX1: Mutasi gen DCX. Berbanding dengan lissencephaly yang disebabkan oleh mutasi LIS1, DCX menunjukkan korteks enam lapisan dan bukannya empat.
- Lissencephaly terpencil tanpa kecacatan genetik lain yang diketahui
- Cobblestone lissencephaly (jenis 2):
- Sindrom Walker-Warburg
- Sindrom Fukuyama
- Penyakit otot, mata dan otak
- Jenis lain tidak boleh diletakkan ke dalam salah satu daripada dua kumpulan di atas:
- LIS2: Sindrom Norman-Roberts, serupa dengan lissencephaly jenis I atau sindrom Miller-Dieker, tetapi tanpa pemadaman kromosom 17.
- LIS3
- LISX2
Microlissencephaly: Ini adalah gabungan ketiadaan lipatan kortikal normal dan kepala kecil yang luar biasa. Kanak-kanak dengan lissencephaly biasa mempunyai kepala bersaiz normal semasa lahir. Kanak-kanak dengan saiz kepala kecil semasa lahir biasanya didiagnosis dengan microlissencephaly.
Ia juga penting untuk membezakan antara lissencephaly dan polymicrogyria, yang merupakan kecacatan perkembangan otak yang berbeza.
Siapa yang hendak dihubungi?
Rawatan lissencephaly
Lissencephaly adalah kecacatan organik yang tidak boleh diubati, jadi hanya rawatan sokongan dan gejala yang mungkin. [ 29 ]
Pertama sekali, ini adalah penggunaan ubat anticonvulsants dan antiepileptik, serta pemasangan tiub gastrostomi di dalam perut (jika kanak-kanak tidak dapat menelan secara bebas). Urut berguna.
Dalam kes hidrosefalus yang teruk, cecair serebrospinal dikeluarkan.
Pencegahan
Pakar mengesyorkan agar ibu bapa masa depan mendapatkan kaunseling genetik, dan wanita hamil mendaftar dengan pakar obstetrik dan ginekologi tepat pada masanya dan menjalani semua peperiksaan yang dijadualkan.
Ramalan
Bagi kanak-kanak dengan lissencephaly, prognosis bergantung pada tahapnya, tetapi selalunya perkembangan mental kanak-kanak tidak melebihi tahap empat hingga lima bulan. Dan semua kanak-kanak dengan diagnosis ini mengalami gangguan psikomotor yang teruk dan epilepsi yang sukar dirawat. [ 30 ]
Menurut NINDS (Institut Gangguan Neurologi dan Strok Kebangsaan Amerika), jangka hayat maksimum bagi penghidap lissencephaly adalah kira-kira 10 tahun.