
Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Polimiositis dan dermatomyositis: punca, gejala, diagnosis, rawatan
Pakar perubatan artikel itu

Ulasan terakhir: 05.07.2025
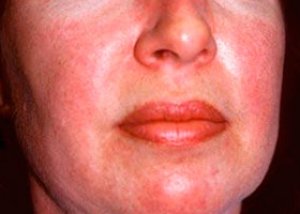
Polymyositis dan dermatomyositis adalah penyakit reumatik sistemik yang jarang berlaku yang dicirikan oleh perubahan keradangan dan degeneratif pada otot (polymyositis) atau otot dan kulit (dermatomyositis). Manifestasi kulit yang paling spesifik ialah ruam heliotrope.
Penglibatan otot adalah simetri dan termasuk kelemahan, sedikit kelembutan, dan atrofi seterusnya otot ikat pinggang pelvis proksimal. Komplikasi mungkin termasuk penglibatan viseral dan keganasan. Diagnosis adalah berdasarkan persembahan klinikal dan penilaian disfungsi otot dengan mengukur tahap enzim, melakukan MRI, elektromiografi, dan biopsi otot. Rawatan melibatkan glukokortikoid, kadangkala digabungkan dengan imunosupresan atau imunoglobulin intravena.
Wanita sakit dua kali lebih kerap daripada lelaki. Penyakit ini boleh berlaku pada mana-mana umur, tetapi paling kerap dikesan dalam lingkungan 40 hingga 60 tahun; pada kanak-kanak - dari 5 hingga 15 tahun.
 [
[ Apakah yang menyebabkan dermatomyositis dan polymyositis?
Punca penyakit itu dipercayai tindak balas autoimun terhadap tisu otot pada individu yang terdedah secara genetik. Penyakit ini lebih biasa dengan kehadiran sejarah keluarga yang terbeban dan pembawa antigen HLA tertentu (DR3, DR52, DR56). Pencetus yang mungkin adalah myositis virus dan neoplasma malignan. Terdapat laporan pengesanan struktur yang serupa dengan picornavirus dalam sel otot; di samping itu, virus boleh menyebabkan penyakit yang sama pada haiwan. Perkaitan tumor malignan dengan dermatomyositis (lebih jarang berbanding dengan polimiositis) menunjukkan bahawa pertumbuhan tumor juga boleh menjadi pencetus kepada perkembangan penyakit akibat daripada permulaan tindak balas autoimun terhadap antigen biasa tumor dan tisu otot.
Deposit IgM, IgG, dan komponen ketiga pelengkap ditemui di dinding saluran darah otot rangka; ini adalah ciri khas dermatomyositis pada kanak-kanak. Pesakit dengan polimiositis juga mungkin mengalami proses autoimun yang lain.
Patofisiologi dermatomyositis dan polimiositis
Perubahan patologi termasuk kerosakan sel dan atrofi terhadap latar belakang keradangan dengan pelbagai tahap keparahan. Otot-otot bahagian atas dan bawah, serta muka, rosak pada tahap yang lebih rendah daripada otot rangka yang lain. Kerosakan pada otot visceral pharynx dan esofagus atas, kurang kerap jantung, perut atau usus, boleh menyebabkan disfungsi organ di atas. Kepekatan myoglobin yang tinggi akibat rhabdomyolysis boleh menyebabkan kerosakan buah pinggang. Perubahan keradangan pada sendi dan paru-paru juga mungkin berlaku, terutamanya pada pesakit yang mempunyai antibodi antisynthetase.
Gejala dermatomyositis dan polimiositis
Permulaan polimiositis mungkin akut (terutamanya pada kanak-kanak) atau subakut (lebih kerap pada orang dewasa). Jangkitan virus akut kadang-kadang mendahului atau mencetuskan manifestasi penyakit, manifestasi yang paling biasa adalah kelemahan otot proksimal atau ruam kulit. Kesakitan dinyatakan pada tahap yang lebih rendah daripada kelemahan. Polyarthralgia, fenomena Raynaud, disfagia, gangguan paru-paru, gejala umum (peningkatan suhu badan, penurunan berat badan, kelemahan) mungkin berkembang. Fenomena Raynaud sering dijumpai pada pesakit dengan penyakit tisu penghubung yang bersamaan.
Kelemahan otot mungkin berkembang selama beberapa minggu atau bulan. Walau bagaimanapun, untuk manifestasi klinikal kelemahan otot, sekurang-kurangnya 50% gentian otot mesti terjejas (oleh itu, kehadiran kelemahan otot menunjukkan perkembangan myositis). Pesakit mungkin mengalami kesukaran untuk mengangkat tangan mereka melebihi paras bahu, berjalan menaiki tangga, atau bangkit dari posisi duduk. Disebabkan kelemahan otot pelvis dan bahu yang teruk, pesakit mungkin terkurung di kerusi roda atau katil. Jika fleksor leher terjejas, menjadi mustahil untuk mengangkat kepala dari bantal. Kemesraan otot-otot faring dan esofagus atas membawa kepada gangguan menelan dan regurgitasi. Otot bahagian bawah, anggota atas dan muka biasanya tidak terjejas. Walau bagaimanapun, kontraktur anggota badan mungkin berkembang.
Ruam kulit yang diperhatikan dalam dermatomyositis biasanya berwarna gelap dan eritematous. Edema periorbital warna ungu (ruam heliotrope) juga merupakan ciri. Ruam kulit mungkin meningkat sedikit di atas paras kulit dan licin atau bersisik; penyetempatan ruam adalah dahi, leher, bahu, dada, belakang, lengan bawah, bahagian bawah tulang kering, kening, kawasan lutut, malleoli medial, permukaan dorsal sendi interphalangeal dan metacarpophalangeal, di sisi sisi (gejala Gottron). Hiperemia pada pangkal atau pinggir kuku adalah mungkin. Dermatitis desquamative, disertai dengan retakan, boleh berkembang pada kulit permukaan sisi jari. Lesi kulit primer selalunya sembuh tanpa sekuela, tetapi boleh menyebabkan perubahan sekunder seperti pigmentasi gelap, atrofi, parut atau vitiligo. Kalsifikasi subkutan mungkin berlaku, terutamanya pada kanak-kanak.
Kira-kira 30% pesakit mengalami polyarthralgia atau polyarthritis, selalunya disertai dengan edema dan efusi sendi. Walau bagaimanapun, keterukan manifestasi sendi adalah kecil. Ia berlaku lebih kerap apabila antibodi kepada Jo-1 atau sintetase lain dikesan pada pesakit.
Penglibatan organ dalaman (kecuali farinks dan esofagus atas) adalah kurang biasa dalam polimiositis berbanding penyakit reumatik lain (terutamanya SLE dan sklerosis sistemik). Jarang, terutamanya dalam sindrom antisynthetase, penyakit ini ditunjukkan sebagai pneumonitis interstisial (dalam bentuk dyspnea dan batuk). Aritmia jantung dan gangguan konduksi mungkin berlaku, tetapi ia biasanya tanpa gejala. Manifestasi gastrousus adalah lebih biasa pada kanak-kanak yang juga mempunyai vaskulitis dan mungkin termasuk muntah dengan darah, melena, dan perforasi usus.
Di mana ia terluka?
Klasifikasi polimiositis
Terdapat 5 jenis polimiositis.
- Polimiositis idiopatik primer, yang boleh berlaku pada sebarang umur, tidak melibatkan kulit.
- Dermatomyositis idiopatik primer adalah serupa dengan polimiositis idiopatik primer tetapi melibatkan kulit.
- Polimiositis dan dermatomyositis yang dikaitkan dengan neoplasma malignan boleh berlaku pada pesakit pada sebarang umur; perkembangan mereka paling kerap diperhatikan pada pesakit tua, serta pada pesakit dengan penyakit tisu penghubung lain. Perkembangan neoplasma malignan boleh diperhatikan dalam tempoh 2 tahun sebelum dan dalam tempoh 2 tahun selepas permulaan myositis.
- Polimiositis kanak-kanak atau dermatomyositis dikaitkan dengan vaskulitis sistemik.
- Polimiositis dan dermatomyositis juga boleh berlaku pada pesakit dengan penyakit tisu penghubung lain, yang paling biasa adalah sklerosis sistemik progresif, penyakit tisu penghubung campuran, dan SLE.
Kemasukan myositis otot truncal dalam kumpulan polymyositis adalah tidak betul, kerana yang terakhir adalah penyakit berasingan yang dicirikan oleh manifestasi klinikal yang serupa dengan polymyositis idiopatik kronik. Walau bagaimanapun, ia berkembang pada usia tua, sering menjejaskan otot bahagian distal badan (contohnya, bahagian atas dan bawah), mempunyai tempoh yang lebih lama, kurang bertindak balas terhadap rawatan, dan dicirikan oleh gambaran histologi yang tipikal.
Diagnosis dermatomyositis dan polimiositis
Polimiositis harus disyaki pada pesakit dengan aduan kelemahan otot proksimal, dengan atau tanpa kelembutan. Penilaian untuk dermatomyositis adalah perlu pada pesakit dengan aduan ruam yang menyerupai heliotrope atau tanda Gottron, serta pada pesakit dengan manifestasi polimiositis yang digabungkan dengan sebarang lesi kulit yang konsisten dengan dermatomyositis. Manifestasi klinikal polimiositis dan dermatomyositis mungkin menyerupai sklerosis sistemik atau, lebih jarang, SLE atau vaskulitis. Kepastian diagnosis meningkat dengan memenuhi sebanyak mungkin lima kriteria berikut:
- kelemahan otot proksimal;
- ruam kulit ciri;
- peningkatan aktiviti enzim tisu otot (creatine kinase atau, jika tiada peningkatan dalam aktivitinya, aminotransferases atau aldolase);
- perubahan ciri dalam miografi atau MRI;
- perubahan histologi ciri dalam biopsi tisu otot (kriteria mutlak).
Biopsi otot boleh mengecualikan beberapa keadaan klinikal yang serupa, seperti myositis pada otot truncal dan rhabdomyolysis akibat jangkitan virus. Perubahan yang didedahkan oleh pemeriksaan histologi mungkin berbeza-beza, tetapi keradangan kronik, fokus degenerasi otot dan penjanaan semula adalah tipikal. Diagnosis yang tepat (biasanya melalui pengesahan histologi) diperlukan sebelum memulakan rawatan yang berpotensi toksik. MRI boleh mengesan fokus edema dan keradangan pada otot, diikuti dengan biopsi yang disasarkan.
Kajian makmal boleh mengesahkan atau, sebaliknya, menghapuskan syak wasangka kehadiran penyakit, dan juga berguna dalam menilai keparahannya, kemungkinan gabungan dengan patologi lain yang serupa dan mendiagnosis komplikasi. Walaupun fakta bahawa antibodi antinuklear dikesan pada sesetengah pesakit, fenomena ini lebih tipikal untuk penyakit tisu penghubung yang lain. Kira-kira 60% pesakit mempunyai antibodi terhadap antigen nukleus (PM-1) atau seluruh sel timus dan Jo-1. Peranan autoantibodi dalam patogenesis penyakit masih tidak jelas, walaupun diketahui bahawa antibodi kepada Jo-1 adalah penanda khusus sindrom antisynthetase, termasuk alveolitis fibrosing, fibrosis pulmonari, arthritis dan fenomena Raynaud.
Penilaian berkala aktiviti creatine kinase berguna untuk memantau rawatan. Walau bagaimanapun, pada pesakit dengan pembaziran otot yang teruk, aktiviti enzim mungkin normal walaupun terdapat myositis aktif kronik. MRI, biopsi otot, atau aktiviti creatine kinase yang tinggi selalunya membantu dalam membezakan antara berulang polimiositis dan miopati yang disebabkan oleh glukokortikoid.
Oleh kerana ramai pesakit mempunyai keganasan yang tidak didiagnosis, sesetengah penulis mengesyorkan agar semua orang dewasa dengan dermatomyositis dan mereka yang mempunyai polimiositis berumur lebih dari 60 tahun menggunakan jadual berikut: pemeriksaan fizikal, termasuk pemeriksaan payudara, pelvis dan rektum (termasuk ujian najis untuk darah ghaib); kiraan darah lengkap; kimia darah; mamografi; ujian antigen karsinoembrionik; urinalisis; radiografi dada. Sesetengah penulis mempersoalkan keperluan untuk pemeriksaan sedemikian pada pesakit yang lebih muda yang tidak mempunyai bukti klinikal keganasan.
Apa yang perlu diperiksa?
Bagaimana untuk memeriksa?
Rawatan dermatomyositis dan polymyositis
Sehingga keradangan lega, aktiviti fizikal harus dihadkan. Glukokortikoid adalah ubat lini pertama. Pada peringkat akut penyakit ini, pesakit dewasa perlu diberi prednisolone (secara lisan) pada dos 40 hingga 60 mg sehari. Penentuan tetap aktiviti creatine kinase adalah penunjuk awal keberkesanan: dalam kebanyakan pesakit, penurunan atau normalisasi dicatatkan dalam masa 6 hingga 12 minggu berikutan peningkatan kekuatan otot. Selepas menormalkan aktiviti enzim, dos prednisolon dikurangkan: pertama dengan kira-kira 2.5 mg sehari selama seminggu, kemudian lebih cepat; jika aktiviti enzim otot meningkat berulang, dos hormon dinaikkan semula. Pesakit yang pulih boleh melakukan tanpa glukokortikoid, tetapi kebanyakan pesakit dewasa memerlukan terapi glukokortikoid jangka panjang (10-15 mg prednisolon setiap hari). Dos awal prednisolone untuk kanak-kanak ialah 30-60 mg/m 2 sekali sehari. Dengan adanya remisi selama > 1 tahun pada kanak-kanak, terapi glukokortikoid boleh dihentikan.
Dalam sesetengah kes, pesakit yang menerima dos glukokortikoid yang tinggi mengalami peningkatan mendadak dalam kelemahan otot, yang mungkin dikaitkan dengan perkembangan miopati glukokortikoid.
Sekiranya tindak balas yang tidak mencukupi terhadap rawatan glukokortikoid, serta dalam kes perkembangan myopathy glucocorticoid atau komplikasi lain yang memerlukan pengurangan dos atau pemberhentian prednisolone, imunosupresan (methotrexate, cyclophosphamide, azathioprine, cyclosporine) harus digunakan. Sesetengah pesakit mungkin menerima hanya methotrexate (biasanya dalam dos yang melebihi dos yang digunakan untuk rawatan RA) selama lebih daripada 5 tahun. Imunoglobulin intravena mungkin berkesan pada pesakit yang refraktori terhadap terapi ubat, tetapi penggunaannya meningkatkan kos rawatan.
Myositis yang dikaitkan dengan tumor primer dan metastatik, serta myositis otot batang, biasanya lebih tahan api terhadap terapi glukokortikoid. Pengampunan myositis yang berkaitan dengan tumor malignan adalah mungkin selepas penyingkiran tumor.
Apakah prognosis untuk dermatomyositis dan polimiositis?
Remisi jangka panjang (dan juga pemulihan klinikal) selama 5 tahun diperhatikan pada lebih separuh daripada pesakit yang dirawat; pada kanak-kanak, angka ini lebih tinggi. Walau bagaimanapun, kambuh semula boleh berlaku pada bila-bila masa. Kadar kelangsungan hidup lima tahun keseluruhan adalah 75%, lebih tinggi pada kanak-kanak. Punca kematian pada orang dewasa adalah kelemahan otot yang teruk dan progresif, disfagia, penurunan pemakanan, pneumonia aspirasi atau kegagalan pernafasan akibat jangkitan paru-paru. Polimiositis lebih teruk dan tahan terhadap rawatan jika terdapat kerosakan pada jantung dan paru-paru. Kematian pada kanak-kanak boleh berlaku akibat vaskulitis usus. Prognosis umum penyakit ini juga ditentukan oleh kehadiran neoplasma malignan.