
Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Xeroderma pigmentosum
Pakar perubatan artikel itu

Ulasan terakhir: 04.07.2025
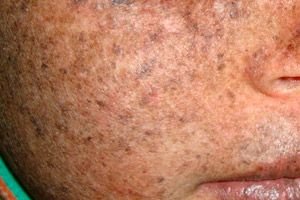
Xeroderma pigmentosum adalah gangguan genetik resesif autosomal pembaikan DNA. Penyakit ini disebabkan oleh mutasi gen yang terlibat dalam pembaikan DNA yang rosak oleh sinaran ultraungu dan bahan toksik lain.
Selalunya, penyakit ini memberi kesan kepada kanak-kanak, yang juga dipanggil "kanak-kanak malam." Komplikasi penyakit yang kerap adalah karsinoma sel basal dan neoplasma malignan lain pada kulit, melanoma malignan metastatik dan karsinoma sel skuamosa.
 [ 1 ]
[ 1 ]
Punca xeroderma pigmentosum
Xeroderma pigmentosum adalah penyakit keturunan, disebarkan oleh gen autosomal daripada ibu bapa, saudara mara dan mempunyai sifat kekeluargaan. Makan tengah hari (1967) dalam satu keluarga tujuh adik beradik menemui tanda-tanda klinikal xeroderma pigmentosum dalam lima daripada mereka.
Patogenesis
Kekurangan enzim endonuklease UV dalam sel pesakit (dalam fibroblas) atau ketiadaan sepenuhnya memainkan peranan penting dalam perkembangan penyakit. Enzim ini bertanggungjawab untuk pembiakan DNA yang rosak oleh sinar ultraviolet. Menurut sumber lain, kekurangan enzim DNA polimerase-1 memainkan peranan penting dalam perkembangan penyakit pada sesetengah pesakit. Penyakit ini paling kerap berkembang di bawah pengaruh sinar dengan panjang gelombang 280-310 nm. Adalah dipercayai bahawa pigmen xeroderma berkembang disebabkan oleh kemasukan pelbagai bahan fotodinamik ke dalam badan atau peningkatan porfirin dalam persekitaran biologi manusia.
Pada peringkat awal penyakit ini, hiperkeratosis, atrofi tumpuan epidermis, penipisan lapisan Malpighian, peningkatan butiran melanin dalam lapisan basal sel, dan keradangan kronik menyusup di lapisan atas dermis (terutamanya di sekitar saluran darah) diperhatikan. Selepas itu, perubahan degeneratif dikesan dalam kolagen dan gentian elastik, dan pada peringkat tumor, tanda-tanda ciri kanser kulit didedahkan.
Gejala xeroderma pigmentosum
Lelaki dan wanita sama-sama terkena penyakit ini. Penyakit ini bermula pada awal kanak-kanak pada musim bunga atau musim panas. Kulit pesakit sangat sensitif terhadap cahaya matahari. Oleh itu, ruam pertama dalam bentuk hiperpigmentasi, serupa dengan tahi lalat, muncul di kawasan kulit yang terdedah kepada cahaya matahari. Ruam meningkat, eritema meningkat, menjadi coklat gelap, telangiectasia, angioma dan atrofi muncul. Selepas itu, papilloma dan ketuat tumbuh (terutamanya pada kulit muka dan leher), bintik-bintik dan ulser muncul. Akibat parut ulser, hidung menjadi lebih nipis ("paruh burung"), kelopak mata tumbuh. Papilloma biasanya berkembang menjadi tumor malignan. Xeroderma berpigmen, sebagai peraturan, berlaku bersama-sama dengan kanser spinoselular, melanosarcomas.
Apa yang mengganggumu?
Tahap
Dalam kursus klinikal xeroderma pigmentosum, lima peringkat dibezakan: keradangan (erythematous), hiperpigmentasi, atrofi, hiperkeratosis dan tumor malignan.
Semasa peringkat keradangan (erythematous), bengkak, bintik merah, dan kadangkala lepuh dan vesikel muncul pada kawasan kulit yang terdedah kepada cahaya matahari (muka, leher, dada atas, lengan, tangan). Hampir tiada ruam pada kawasan yang tidak terdedah kepada cahaya matahari.
Dengan hiperpigmentasi, coklat muda, bintik-bintik hiperpigmen coklat yang serupa dengan tahi lalat muncul sebagai ganti bintik merah.
Dengan atrofi, kulit menjadi kering, menjadi lebih nipis, dan berkedut. Banyak telangiektasia kecil, stellate dan parut dengan permukaan berkilat diperhatikan pada kulit bibir dan hidung. Disebabkan oleh atrofi dan parut, mikrotomi (pengurangan pembukaan mulut), ektropion, penipisan telinga dan hidung, atresia pembukaan hidung dan mulut berkembang. Dalam 80-85% pesakit, mata terjejas: konjunktivitis, keratitis, kerosakan pada kornea dan membran mukus, dan kelemahan penglihatan diperhatikan. Dyschromia, telangiectasia, perubahan hiperkeratotik dan tumor diperhatikan pada kulit kelopak mata.
Dengan hiperkeratosis, tumor ketuat, papilloma, keratoacanthoma, fibroma, angiofibrioma dan tumor jinak lain muncul dalam fokus patologi yang diterangkan di atas. Oleh itu, sesetengah saintis memasukkan pigmen xeroderma dalam kumpulan penyakit prakanser.
Selepas 10-15 tahun dari permulaan penyakit, tumor kulit malignan (basalioma, endothelioma, angiosarcoma) muncul dalam foci dan bintik pigmen yang diubah secara atropik. Tumor dimusnahkan dalam masa yang singkat, bermetastasis ke organ dalaman dan membawa kepada kematian. Dalam organ dan tisu dalaman sesetengah pesakit, perubahan distrofik umum diperhatikan (syndactelia jari kaki kedua dan ketiga, distrofi pergigian, keguguran rambut lengkap, dll.).
Borang
Bentuk neurologi xeroderma pigmentosum menunjukkan dirinya dalam dua sindrom.
Sindrom Reed dicirikan oleh manifestasi klinikal xeroderma pigmentosum dan microcephaly, idiopati, dan pertumbuhan perlahan sistem rangka. Dalam bentuk neurologi penyakit, pembaikan DNA adalah sukar dan terapi sinar-X membawa kepada peningkatan dalam fokus patologi utama.
Sindrom De Sinctis-X Cocchione dicirikan oleh tanda-tanda klinikal berikut:
- perkembangan tanda-tanda klinikal xeroderma pigmentosum pada kulit terutamanya fotosensitif;
- penampilan awal tumor malignan;
- kursus serentak lumpuh spastik, mikrosefali dan demensia;
- kecacatan kongenital dan kerdil;
- kurang pembangunan gonad;
- kerap keguguran;
- penghantaran resesif melalui warisan.
Menurut beberapa pakar dermatologi, sindrom Santis-Cacchione bukanlah penyakit bebas, tetapi manifestasi klinikal xeroderma pigmentosum yang teruk dan dinyatakan sepenuhnya.
Bentuk genetik
Jenis |
Gen |
Lokus |
Penerangan |
Jenis A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) kumpulan A - bentuk klasik |
Jenis B, II, XPB |
XPB |
2q21 |
XP Kumpulan B |
Jenis C, III, XPC |
XPC |
3p25 |
XP Kumpulan C |
Jenis D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Kumpulan D atau Sindrom De Sanctis-Cacchione |
Jenis E, V, XPE |
DDB2 |
11p12-p11 |
XP Kumpulan E |
Jenis F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Kumpulan F |
Jenis G, VII, XPG |
RAD2ERCC5 |
13q33 |
Kumpulan XP G dan sindrom COFS (sindrom cerebro-oculo-facioskeletal) jenis 3 |
Jenis V, XPV |
POLH |
6p21.1-p12 |
Varian Xeroderma Pigmentosa |
Apa yang perlu diperiksa?
Bagaimana untuk memeriksa?
Siapa yang hendak dihubungi?
Rawatan xeroderma pigmentosum
Ubat antimalaria (delagyl, plaquenil, resoquin, dll.), melindungi DNA daripada sinaran ultraungu, menghalang proses penyahpolimeran, mengurangkan kepekaan (fotosensitisasi) kulit kepada cahaya matahari. Ubat-ubatan ini mempunyai sifat anti-radang dan hyposensitizing. Adalah disyorkan untuk menjalankan terapi am bersama-sama dengan terapi vitamin (B1, B2, PP, B6, B12, A, E), antihistamin (tavegil, diphenhydramine, suprastin), agen desensitisasi (natrium tiosulfat, 10% kalsium klorida secara intravena 10 ml).
Untuk rawatan tempatan, krim pelindung matahari dan salap digunakan.
Dalam bentuk tumor xeroderma pigmen, kaedah pembedahan, nitrogen cecair dan sinar laser digunakan. Untuk melindungi diri anda daripada cahaya matahari, anda mesti memakai pakaian longgar, topi matahari dan sarung tangan.
Ramalan
Kebanyakan pesakit (2/3) meninggal dunia sebelum mencapai umur 15 tahun. Sesetengah pesakit boleh hidup hingga 40-50 tahun.
[ 20 ]