
Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Sindrom Angelman pada kanak-kanak dan orang dewasa
Pakar perubatan artikel itu

Ulasan terakhir: 04.07.2025

Terdapat beberapa penyakit yang mana ungkapan seperti "jaga diri anda dan anda tidak akan sakit" bunyi, sekurang-kurangnya, tidak masuk akal. Ini adalah patologi di mana beberapa keabnormalan mental dan fizikal wujud dalam tubuh kanak-kanak walaupun sebelum kelahiran, tetapi ibu bapa tidak boleh dipersalahkan untuk ini. Penyakit sedemikian disebabkan oleh mutasi atau kelainan pada set kromosom dan dipanggil kromosom atau genetik. Sindrom Angelman, Sindrom Down, Sindrom Patau, Sindrom Edwards, Sindrom Turner, Sindrom Prader-Willi - ini hanya sebahagian daripada penyakit genetik dari senarai yang agak baik.
Sindrom Lelaki Bahagia
Kali ini kita akan bercakap tentang patologi yang dinamakan sempena pakar kanak-kanak Inggeris Harry Angelman, yang pertama kali membangkitkan isu masalah ini pada tahun 1965, setelah menemui tiga kanak-kanak yang luar biasa dalam amalannya sehari sebelumnya, disatukan oleh gejala pelik yang biasa. Doktor memanggil kanak-kanak ini sebagai anak patung dan menulis artikel tentang mereka, yang pada mulanya dipanggil "Children-marionettes". Artikel itu sendiri dan tajuknya ditulis di bawah kesan lukisan yang dilihat di salah satu muzium Verona. Lukisan itu menggambarkan seorang budak lelaki ketawa, dan ia dipanggil "The Puppet Boy". Pergaulan kanak-kanak yang digambarkan dalam lukisan itu dengan tiga kanak-kanak yang pernah ditemui Angelman dalam amalannya mendorong pakar pediatrik untuk menggabungkan kanak-kanak itu ke dalam satu kumpulan kerana penyakit yang mereka hadapi.
Tidak ada yang menghairankan bahawa kanak-kanak yang disebutkan dalam artikel itu tidak disedari oleh doktor lain. Lagipun, pada pandangan pertama nampaknya mereka mempunyai penyakit yang sama sekali berbeza, jadi berbeza adalah gambaran klinikal umum penyakit itu dalam 3 kes yang berbeza. Mungkin patologi kromosom "baru" akan menarik minat saintis lain, tetapi pada masa itu genetik belum cukup berkembang untuk mengesahkan hipotesis doktor Inggeris. Oleh itu, selepas minat tertentu di dalamnya, artikel itu dibuang di rak belakang untuk masa yang lama.
Sebutan seterusnya mengenai sindrom Angelman, yang merupakan artikel oleh pakar pediatrik Inggeris G. Angelman yang kini dipanggil, bermula pada awal 80-an abad ke-20. Dan hanya pada tahun 1987 adalah mungkin untuk mencari sebab mengapa sebahagian kecil kanak-kanak dilahirkan dengan penyimpangan sedemikian rupa sehingga dari luar mereka kelihatan sentiasa tersenyum dan gembira. Sebenarnya, ini tidak benar sama sekali, dan senyuman itu hanyalah sebuah renungan, di belakangnya terselit jiwa manusia yang tidak bahagia dan keperitan ibu bapa.
Epidemiologi
Menurut statistik, mutasi kromosom pada kanak-kanak boleh berkembang dengan latar belakang mutasi yang serupa pada ibu bapa dan jika tiadanya. Tidak ada sifat keturunan yang jelas dari sindrom Angelman (AS), tetapi kebarangkalian untuk membangunkan patologi pada ibu bapa dengan mutasi kromosom agak tinggi.
Menarik juga, jika sesebuah keluarga sudah mempunyai anak dengan AS, terdapat satu peratus peluang untuk mendapat anak kedua dengan gangguan yang sama, walaupun ibu bapanya sihat.
Masih tiada statistik tepat mengenai bilangan pesakit dengan sindrom Angelman. Mungkin sebabnya adalah pelbagai gejala, yang mungkin berlaku dalam komposisi tertentu atau tidak berlaku sama sekali untuk masa yang lama. Diandaikan bahawa kelaziman penyakit ini ialah: 1 kanak-kanak setiap 20,000 bayi baru lahir. Tetapi angka ini sangat anggaran.
Punca Sindrom Angelman
Sindrom Angelman adalah nama perubatan untuk patologi kromosom, tetapi ia jauh dari satu-satunya. Orang panggil sindrom anak patung penyakit ini, sindrom boneka gembira, sindrom Petrushka, dan sindrom anak patung ketawa. Orang datang dengan pelbagai nama (kadang-kadang menyinggung perasaan pesakit itu sendiri dan ibu bapa mereka), tetapi penyakit adalah penyakit, tidak kira betapa lucunya ia mungkin kelihatan dan tidak kira apa sebabnya.
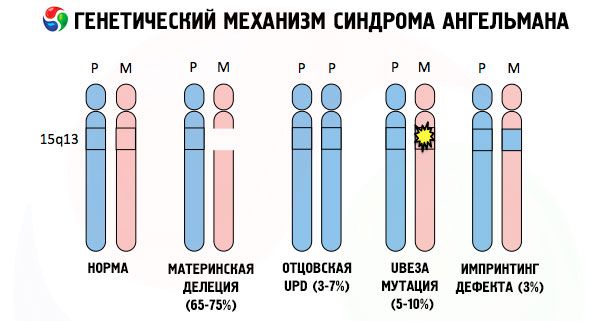
Dan sebab-sebab perkembangan sindrom Angelman, seperti banyak patologi genetik lain, dalam semua kes adalah gangguan dalam struktur salah satu kromosom atau set kromosom secara keseluruhan. Tetapi dalam kes kami, keseluruhan masalah terletak pada kromosom 15, yang diturunkan dari ibu. Iaitu, kromosom bapa dalam kes ini tidak mempunyai penyimpangan, tetapi wanita mengalami mutasi tertentu.
Mengikut jenis kelainan kromosom, sindrom Angelman dikelaskan sebagai mutasi kromosom. Mutasi sedemikian dianggap sebagai:
- Pemadaman (ketiadaan bahagian kromosom yang mengandungi set gen tertentu; jika salah satu gen hilang, kita bercakap tentang pemadaman mikro), yang merupakan hasil daripada dua pecah dan satu penyatuan semula, apabila bahagian kromosom asal hilang.
- Penduaan (kehadiran bahagian tambahan dalam kromosom yang merupakan salinan yang sedia ada), yang dalam kebanyakan kes membawa kepada kematian seseorang, dan kurang kerap kepada ketidaksuburan.
- Penyongsangan (pembalikan salah satu bahagian kromosom sebanyak 180 darjah, iaitu dalam arah yang bertentangan, dan kemudian gen di dalamnya terletak dalam susunan yang bertentangan), apabila hujung kromosom yang patah disambungkan dalam susunan yang berbeza daripada asal.
- Sisipan (jika sebahagian daripada bahan genetik dalam kromosom tidak pada tempatnya),
- translokasi (jika bahagian tertentu kromosom dilekatkan pada kromosom lain; mutasi sedemikian boleh berlaku bersama tanpa kehilangan bahagian).
Menerima kromosom bermutasi daripada ibu yang tidak curiga, kanak-kanak itu ditakdirkan untuk dilahirkan dengan kelainan. Penyebab paling biasa sindrom Angelman masih dianggap sebagai penghapusan kromosom ke-15 ibu, apabila bahagian kecil hilang. Mutasi yang kurang biasa dalam sindrom "anak patung ketawa" dianggap sebagai:
- translokasi,
- disomi unipaternal (jika anak menerima sepasang kromosom daripada bapa, kromosom ibu tidak hadir),
- mutasi gen dalam DNA, yang merupakan bahan binaan utama (genetik) dan arahan untuk penggunaannya yang betul (khususnya, mutasi gen ube3a dalam kromosom ibu).
Kehadiran salah satu mutasi ini pada ibu bapa adalah faktor risiko untuk perkembangan sindrom Angelman pada kanak-kanak. Tetapi bukan sahaja mutasi kromosom, tetapi juga genomik (yang dikaitkan dengan perubahan kuantitatif dalam set kromosom dan lebih biasa daripada kromosom) boleh mencetuskan perkembangan penyakit pada kanak-kanak. Mutasi genomik biasa termasuk trisomi kromosom (jika set kromosom seseorang mempunyai lebih daripada 46 kromosom).
Untuk patologi muncul pada kanak-kanak, ibu bapa sama sekali tidak perlu mempunyai kelainan kromosom. Namun, terdapat peratusan tertentu pesakit yang penyakitnya adalah keturunan.
Patogenesis
Mari kita mendalami sedikit tentang biologi, atau lebih tepat lagi, genetik. Maklumat genetik setiap organisma manusia terkandung dalam 23 pasang kromosom. Satu kromosom daripada sepasang diteruskan kepada anak daripada bapa, satu lagi daripada ibu. Semua pasangan kromosom berbeza dalam bentuk dan saiz dan membawa maklumat tertentu. Oleh itu, pasangan kromosom ke-23 (kromosom X dan Y) bertanggungjawab untuk pembentukan ciri seksual bayi (XX - perempuan, XY - lelaki, manakala kromosom Y hanya boleh diterima oleh anak daripada bapa).
Sebaik-baiknya, seorang kanak-kanak menerima 46 kromosom daripada ibu bapanya, yang membentuk ciri genetiknya, yang menentukannya sebagai individu. Bilangan kromosom yang lebih besar dipanggil trisomi dan dianggap sebagai penyelewengan daripada norma. Sebagai contoh, kehadiran kromosom 47 dalam set kromosom (karyotype, menentukan spesies dan ciri individu) menyebabkan berlakunya sindrom Down.
Jika kromosom diwarnai dengan pewarna khas, maka di bawah mikroskop anda dapat melihat jalur warna yang berbeza di sepanjang setiap daripada mereka. Di dalam setiap jalur terdapat sejumlah besar gen. Semua jalur ini dinomborkan oleh saintis dan mempunyai lokasi tetap. Ketiadaan salah satu jalur dianggap sebagai penyelewengan dari norma. Dalam sindrom Angelman, seseorang sering dapat memerhatikan ketiadaan segmen kromosom ibu dalam selang q11-q13, terletak di lengan panjang, bilangan pangkalan DNA di mana hanya kira-kira 4 juta.
Komponen utama kromosom dianggap sebagai molekul DNA yang sangat panjang yang mengandungi ribuan gen dan puluhan dan ratusan juta bes nitrogen. Oleh itu, kromosom 15, yang bertanggungjawab untuk perkembangan sindrom Angelman dan beberapa yang lain, mengandungi 1200 gen dan kira-kira 100 juta asas. Sebarang gangguan dalam struktur molekul DNA pastinya akan menjejaskan penampilan dan perkembangan anak masa depan.
Maklumat genetik yang terkandung dalam gen ditukar kepada protein atau RNA. Proses ini dipanggil ekspresi gen. Dengan cara ini, maklumat genetik yang diterima daripada ibu bapa menerima kedua-dua bentuk dan kandungan, yang terkandung dalam waris perempuan atau lelaki mereka yang unik.
Terdapat beberapa patologi dengan jenis warisan bukan klasik, termasuk sindrom Angelman, di mana gen yang diterima daripada ibu bapa sebagai sebahagian daripada kromosom berpasangan mempunyai kesan unik ibu bapa dan menunjukkan diri mereka dalam cara yang berbeza.
Jadi, sindrom Angelman adalah contoh yang menarik bagi pencetakan genomik, di mana ekspresi gen dalam badan kanak-kanak bergantung secara langsung pada ibu bapa mana alel diterima (bentuk berbeza satu gen, diterima daripada bapa dan ibu, terletak pada bahagian yang sama pada kromosom berpasangan). Iaitu, hanya anomali dalam kromosom ibu membawa kepada perkembangan sindrom, manakala mutasi dan gangguan struktur kromosom bapa menyebabkan patologi yang sama sekali berbeza.
Dalam patologi ini, terdapat kekurangan gen tertentu dalam kromosom ibu atau kehilangan/pengurangan dalam aktiviti gen individu (dalam kebanyakan kes, gen ube3a, yang terlibat dalam metabolisme ubiquitin, protein yang mengawal degradasi protein lain). Akibatnya, kanak-kanak itu didiagnosis dengan keabnormalan perkembangan mental dan kecacatan fizikal.
Gejala Sindrom Angelman
Gejala sindrom Angelman menjejaskan pelbagai aspek kehidupan dan perkembangan kanak-kanak: fizikal, neurologi, mental. Berdasarkan ini, 3 kumpulan gejala boleh dikenal pasti yang menunjukkan perkembangan patologi ini.
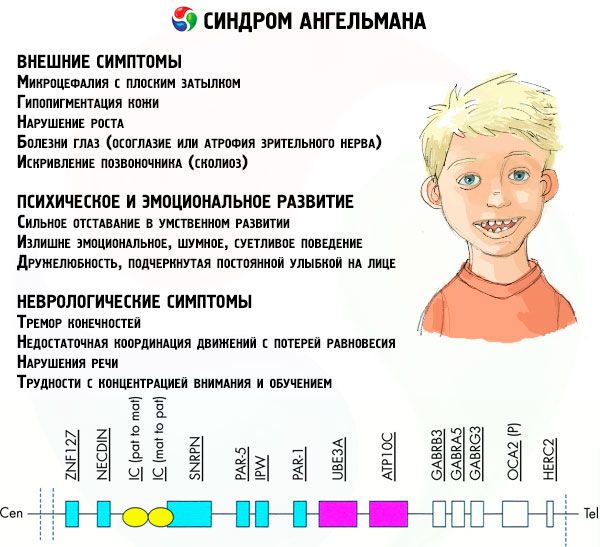
- Gejala luaran atau fizikal:
- kepala yang tidak seimbang berbanding dengan badan dan anggota badan, yang bersaiz normal,
- mulut terlalu lebar,
- hampir selalu ada senyuman di muka (dengan mulut terbuka),
- gigi yang jarang,
- bibir atas yang sempit,
- lidah lebar yang kerap terjulur,
- rahang bawah menonjol,
- dagu runcing,
- kulit yang sangat ringan, selalunya rambut (albinisme, dikaitkan dengan fakta bahawa badan tidak menghasilkan pigmen melanin),
- bintik-bintik gelap pada kulit cerah (hipopigmentasi akibat pengeluaran melanin yang tidak mencukupi)
- gejala fizikal atau luaran: penyakit mata seperti strabismus atau atrofi saraf optik,
- kelengkungan tulang belakang (scoliosis),
- kaki kaku (apabila berjalan, seseorang tidak membengkokkan kakinya di lutut kerana pergerakan sendi yang rendah, oleh itu perbandingan dengan gaya berjalan anak patung).
- Gejala yang berkaitan dengan perkembangan mental dan emosi:
- terencat akal yang teruk,
- tingkah laku yang terlalu emosi, bising, cerewet,
- kerap bertepuk tangan,
- menyatakan keramahan, ditekankan oleh senyuman yang berterusan di wajah,
- kerap ketawa tanpa sebab.
- Gejala neurologi:
- gegaran anggota badan,
- penyelarasan pergerakan yang tidak mencukupi dengan kehilangan keseimbangan,
- mengurangkan nada otot,
- pelbagai gangguan tidur,
- sawan histeria yang kerap pada zaman kanak-kanak,
- gangguan pertuturan (kanak-kanak mula bercakap lewat, mempunyai kemahiran komunikasi yang lemah dan pertuturan yang tidak jelas),
- hiperaktif terhadap latar belakang peningkatan kegembiraan,
- kesukaran menumpukan perhatian dan pembelajaran.
Tetapi ini adalah gambaran umum penyakit ini. Malah, gambaran klinikal sindrom Angelman sebahagian besarnya bergantung pada peringkat perkembangan penyakit dan jenis mutasi kromosom yang menyebabkan patologi. Ini bermakna bahawa gejala penyakit boleh berbeza dengan ketara pada pesakit yang berbeza, yang untuk masa yang lama tidak membenarkan kita membezakan patologi dari orang lain dengan gambaran klinikal yang sama.
Di antara jumlah simptom, kita boleh menyerlahkan ciri-ciri semua pesakit tanpa pengecualian:
- terencat akal yang teruk,
- tingkah laku yang tidak sesuai (ketawa yang tidak munasabah, peningkatan keseronokan, tumpuan yang lemah, keadaan euforia),
- kurang perkembangan kemahiran motor,
- koordinasi pergerakan yang lemah, gaya berjalan ataxia (kelajuan tidak sekata, bergoyang dari sisi ke sisi, dll.), gegaran anggota badan.
- gangguan perkembangan pertuturan dengan penguasaan cara komunikasi bukan lisan.
Antara simptom yang dihadapi oleh kebanyakan pesakit, yang berikut boleh dibezakan:
- ketidakseimbangan antara kepala dan badan yang disebabkan oleh perkembangan fizikal yang lambat,
- dalam kebanyakan pesakit bentuk tengkorak adalah sedemikian rupa sehingga saiz otak kekal lebih kecil daripada orang yang sihat (microcephaly),
- sawan epilepsi sebelum umur 3 tahun dengan penurunan progresif dalam kekuatan dan kekerapan pada usia yang lebih tua,
- herotan parameter EEG (turun naik dan amplitud tinggi gelombang frekuensi rendah).
Gejala ini agak biasa, bagaimanapun, 20% pesakit dengan sindrom Angelman tidak mengalaminya.
Walaupun kurang kerap, adalah mungkin untuk mendiagnosis manifestasi penyakit seperti:
- strabismus teruk atau ringan,
- kawalan pergerakan lidah yang lemah, menyebabkan pesakit sering menjelirkan lidah mereka tanpa sebab,
- kesukaran menelan dan menghisap, terutamanya pada kanak-kanak kecil,
- gangguan pigmentasi kulit dan mata,
- tangan diangkat atau dibengkokkan semasa berjalan,
- hiperrefleksia,
- gangguan tidur, terutamanya pada zaman kanak-kanak,
- air liur yang kerap,
- dahaga yang tidak pernah puas,
- pergerakan mengunyah yang terlalu aktif,
- hipersensitiviti kepada haba,
- belakang kepala rata,
- rahang bawah menonjol,
- tapak tangan licin.
Peratusan besar pesakit mempunyai masalah dengan kencing, yang mereka kurang mengawal, gangguan kemahiran motor halus, yang mewujudkan kesukaran dalam penjagaan diri dan pembelajaran, dan berat badan berlebihan. Hampir semua pesakit mengalami akil baligh lewat daripada rakan sebaya yang sihat.
Kanak-kanak dengan sindrom Angelman memahami ucapan lisan dengan baik dan memahaminya, tetapi tidak mahu mengambil bahagian dalam perbualan, mengehadkan ucapan mereka kepada beberapa dozen perkataan yang diperlukan dalam kehidupan seharian. Walau bagaimanapun, pada masa dewasa, pesakit sedemikian kelihatan lebih muda daripada rakan sebaya mereka tanpa patologi genetik.
Banyak gejala sindrom Angelman tidak tetap, jadi gambaran klinikal penyakit itu berubah dengan ketara dengan usia. Kejang dan sawan epilepsi menjadi kurang kerap atau hilang sama sekali, pesakit menjadi kurang bersemangat, dan tidur bertambah baik.
Komplikasi dan akibatnya
Sindrom Angelman adalah patologi kromosom yang teruk, pada masa ini hampir tidak dapat diubati yang menghalang pesakit daripada peluang untuk menjalani kehidupan normal. Bagaimana kehidupan kanak-kanak dengan AS bergantung pada jenis keabnormalan kromosom.
Penduaan segmen kromosom tidak serasi dengan kehidupan dalam kebanyakan kes. Dan walaupun pesakit sedemikian tidak mati semasa bayi dan mencapai akil baligh, mereka tidak mempunyai peluang untuk mempunyai anak.
Penghapusan atau ketiadaan sebahagian daripada gen yang paling kerap berlaku dalam sindrom Angelman adalah penghalang kepada kanak-kanak belajar berjalan dan bercakap. Kanak-kanak sedemikian mempunyai bentuk terencat mental yang lebih teruk, dan sawan epilepsi berlaku lebih kerap, dan keamatan mereka jauh lebih besar daripada pesakit dengan kelainan kromosom lain.
Sekiranya hanya terdapat mutasi satu gen, dengan perhatian dan pendekatan yang sewajarnya kanak-kanak itu boleh diajar asas penjagaan diri, komunikasi dan interaksi dalam kumpulan, walaupun dia masih akan ketinggalan di belakang rakan sebayanya dalam perkembangan.
Bagi kanak-kanak sindrom Angelman yang bersifat baik hati, yang paling penting ialah kasih sayang dan perhatian ibu bapa mereka. Hanya dalam hal ini pendidikan anak akan membuahkan hasil, walaupun kecil. Sudah tentu, pesakit dengan AS tidak akan dapat belajar di sekolah biasa. Mereka memerlukan kelas khas di mana kanak-kanak mula-mula akan diajar untuk menumpukan perhatian, dan kemudian secara beransur-ansur akan diberikan asas pengetahuan sekolah.
Diagnostik Sindrom Angelman
Sindrom Angelman adalah patologi perkembangan kongenital. Tetapi disebabkan oleh keadaan tertentu, selalunya mustahil untuk mendiagnosisnya pada masa bayi dan awal kanak-kanak. Ini disebabkan oleh ekspresi simptom yang tidak spesifik dan lemah pada bayi dan kanak-kanak di bawah umur 3 tahun. Dan kelaziman penyakit di negara kita tidak begitu hebat sehingga doktor telah belajar mengenalinya di kalangan rakan sebayanya.
Sindrom Angelman pada bayi boleh menampakkan dirinya sebagai penurunan nada otot, yang menampakkan dirinya dalam masalah dengan pemakanan (kelemahan refleks menghisap dan menelan), dan kemudiannya kesukaran belajar berjalan (kanak-kanak seperti itu mula berjalan lebih lama lagi). Gejala-gejala ini adalah tanda-tanda pertama kelainan perkembangan pada bayi, yang mungkin dikaitkan dengan kelainan kromosom. Hanya analisis genetik boleh mengesahkan andaian ini.
Perhatian khusus diberikan kepada kanak-kanak yang ibu bapanya mempunyai pelbagai gangguan genomik atau kromosom. Lagipun, penyakit itu mungkin tidak nyata pada mulanya, dan jika patologi dikesan dalam masa, dengan mula bekerja secara intensif dengan kanak-kanak itu, adalah mungkin untuk mencapai kejayaan yang lebih besar dalam pembelajaran, melambatkan perkembangan penyakit.
Sekiranya ibu bapa mempunyai pelbagai keabnormalan kromosom, analisis genetik dijalankan sebelum bayi dilahirkan, kerana SA adalah salah satu patologi yang dapat dikesan pada peringkat embrio.
Pengumpulan bahan untuk penyelidikan genetik boleh dilakukan dengan dua cara:
- invasif (dengan peratusan risiko tertentu, kerana perlu menembusi rahim untuk mengambil sampel cecair amniotik),
- bukan invasif (analisis DNA bayi daripada darah ibu).
Kajian berikut kemudiannya dijalankan:
- penghibridan in situ pendarfluor (kaedah FISH) – pengikatan probe DNA yang dilabelkan dengan pewarna khas pada DNA yang sedang dikaji, diikuti dengan pemeriksaan di bawah mikroskop.
- analisis mutasi dalam gen ube3a dan gen yang dicetak,
- Analisis metilasi DNA menggunakan kaedah khas yang digunakan dalam genetik.
Ujian genetik memberikan maklumat yang agak tepat dalam kes keabnormalan kromosom, yang bermaksud ibu bapa masa depan tahu terlebih dahulu apa yang perlu disediakan. Walau bagaimanapun, terdapat pengecualian. Dalam kumpulan pesakit tertentu, dengan kehadiran semua gejala yang menunjukkan patologi, keputusan ujian tetap normal. Iaitu, patologi hanya boleh dikenal pasti dengan memerhatikan kanak-kanak dengan teliti dari awal kanak-kanak: bagaimana dia makan, apabila dia mula berjalan dan bercakap, sama ada dia membengkokkan kakinya ketika berjalan, dsb.
Sebagai tambahan kepada kaedah FISH, antara kaedah diagnostik instrumental untuk sindrom Angelman, seseorang boleh memilih tomografi (CT atau MRI), yang membantu menentukan keadaan dan saiz otak, dan electroencephalogram (EEG), yang menunjukkan bagaimana bahagian individu otak berfungsi.
Doktor biasanya membuat diagnosis akhir pada usia 3-7 tahun, apabila pesakit sudah mempunyai kebanyakan gejala dan dinamik perkembangan penyakit dapat dilihat.
Ujian apa yang diperlukan?
Diagnosis pembezaan
Sindrom Angelman adalah patologi genetik yang hampir tidak mempunyai manifestasi khusus. Kebanyakan gejala boleh sama-sama menunjukkan kedua-dua AS dan patologi genetik lain.
Diagnosis pembezaan sindrom Angelman dijalankan dengan patologi berikut:
- Sindrom Pitt-Hopkins (pesakit dicirikan oleh keterbelakangan mental, watak ceria, tersenyum, mereka mempunyai mulut yang agak besar dan lebar, microcephaly diperhatikan). Perbezaannya ialah serangan hiperventilasi dan menahan nafas dalam keadaan terjaga.
- Sindrom Christianson (pesakit adalah orang yang terencat akal dengan perwatakan ceria, tidak boleh bercakap, dicirikan oleh microcephaly, ataxia, sawan, pergerakan otot yang tidak disengajakan).
- Sindrom Mowat-Wilson (gejala: terencat akal, sawan epilepsi, dagu runcing, mulut terbuka, ekspresi gembira di muka, mikrosefali). Perbezaan: jarak yang besar antara mata, mata senget ke dalam, hujung hidung yang membulat, auricle yang dipusing ke belakang.
- Sindrom Kabuki (dicirikan oleh terencat akal ringan hingga sederhana, masalah pertuturan dan motor, kelemahan otot, sawan epilepsi, mikrosefali, selang panjang antara gatal-gatal, dan gangguan koordinasi). Dicirikan oleh kening melengkung, bahagian sisi kelopak mata bawah yang melengkung, mata lebar, fisur palpebra yang panjang dengan bulu mata yang panjang dan tebal.
- Sindrom Rett (pembezaan daripada AS pada wanita). Gejala: perkembangan pertuturan yang lambat, sawan, mikrosefali. Perbezaannya adalah bahawa tidak ada ekspresi gembira di wajah, terdapat serangan apnea dan apraxia, yang berkembang dari masa ke masa.
- Sindrom kelewatan mental resesif autosomal 38 (gejala: terencat akal yang ketara dengan kelewatan dalam kemahiran motor dan pertuturan, kelemahan otot, masalah pemakanan pada peringkat bayi, impulsif). Ciri yang membezakan ialah warna biru pada iris.
- Sindrom pertindihan gen MECP 2 (pembezaan daripada SA pada lelaki). Gejala: terencat akal yang teruk, kelemahan otot sejak kecil, masalah pertuturan atau kekurangan pertuturan, epilepsi. Perbezaan: myopathy progresif, jangkitan berulang yang berterusan.
- Sindrom Kleefstra (gejala: masalah pertuturan dan pemikiran, kelemahan otot, gangguan tidur, kurang perhatian, mulut terbuka, hiperaktif, sawan, ataxia, gangguan keseimbangan). Ciri tersendiri: muka rata, hidung mancung pendek, mata lebar, bibir bawah melengkung besar, letusan agresif.
- Sindrom Smith-Magenis (dicirikan oleh sawan, masalah tidur, gangguan perkembangan intelektual dan motor). Ciri tersendiri termasuk muka yang lebar dan rata serta dahi yang menonjol.
- Sindrom Koolen-de Vries (rencat akal ringan hingga sederhana, kelemahan otot, sawan, keramahan). Ciri yang membezakan: muka panjang dengan dahi tinggi, telinga menonjol, mata senget, mobiliti sendi tinggi, kecacatan jantung kongenital.
- Sindrom Phelan-McDermid (gejala: terencat akal, gangguan pertuturan atau kekurangan pertuturan). Perbezaan: tangan besar dengan otot yang berkembang, kelemahan otot sejak lahir, berpeluh lemah.
Patologi seperti kekurangan adenil succinate, sindrom terencat akal resesif autosomal 1, sindrom duplikasi kromosom 2q23.1, sindrom haploinsufficiency gen FOXG1, STXBP1 atau MEF2C dan beberapa yang lain boleh "megah" dengan simptom yang serupa dengan sindrom Angelman.
Tugas doktor adalah untuk membuat diagnosis yang tepat, membezakan sindrom Angelman daripada patologi dengan gejala yang sama, dan menetapkan rawatan berkesan yang berkaitan dengan peringkat penyakit yang didiagnosis.
Siapa yang hendak dihubungi?
Rawatan Sindrom Angelman
Sindrom Angelman adalah salah satu daripada patologi yang mana ubat masih mencari rawatan yang berkesan. Rawatan etiologi penyakit ini berada dalam peringkat pembangunan pelbagai kaedah dan cara, kebanyakannya belum diuji pada manusia. Ini bermakna bahawa buat masa ini doktor perlu menghadkan diri mereka kepada terapi gejala, yang membantu entah bagaimana mengurangkan keadaan yang tidak menyenangkan kanak-kanak dan orang dewasa dengan sindrom marionette, mengalami sawan epilepsi, air liur, hipotensi dan gangguan tidur.
Oleh itu, adalah mungkin untuk mengurangkan kekerapan dan kekuatan sawan epilepsi dengan bantuan ubat antikonvulsan yang dipilih dengan betul. Tetapi keseluruhan kesukaran adalah bahawa sawan pada pesakit dengan SA berbeza daripada sawan epilepsi biasa kerana ia dicirikan oleh beberapa jenis sawan, yang bermaksud bahawa keadaan itu boleh dikurangkan dengan memberikan beberapa ubat sekaligus.
Antikonvulsan yang paling popular digunakan untuk merawat sindrom Angelman ialah: asid valproik, topiramate, lamotrigine, levetiracetam, clonazepam dan ubat-ubatan berdasarkannya. Kurang biasa digunakan adalah ubat berdasarkan carmazepine, phenytoin, phenobarbital, ethosuximide, kerana sesetengah daripada mereka boleh mencetuskan kesan paradoks yang terdiri dalam menguatkan dan meningkatkan kekerapan sawan epilepsi. Ini berlaku jika ubat digunakan sebagai sebahagian daripada monoterapi.
Untuk merawat air liur, dua kaedah biasanya digunakan: perubatan (ubat yang menyekat pengeluaran air liur) dan pembedahan, yang melibatkan implantasi semula saluran air liur. Tetapi dalam kes SA, kaedah ini dianggap tidak berkesan, dan isu itu tetap terbuka. Ibu bapa dan mereka yang menjaga pesakit sedemikian perlu memberi perhatian khusus kepada isu ini, kerana pesakit itu sendiri biasanya tidak mengawal air liur, dan ada yang tidak dapat menjaga diri mereka sendiri.
Masalah lain ialah tempoh tidur yang pendek. Selalunya kanak-kanak dengan sindrom Angelman tidur tidak lebih daripada 5 jam, yang mempunyai kesan negatif terhadap fungsi seluruh badan. Kanak-kanak yang mudah teruja dan aktif yang suka permainan dan komunikasi (walaupun mereka cuba mengehadkan diri mereka kepada kaedah bukan lisan) kelihatan letih pada siang hari. Untuk mendapatkan rehat yang baik, badan memerlukan tidur yang nyenyak dan penuh, tetapi ini adalah tangkapan yang tepat.
Nampaknya ubat sedatif (phenothiazines dan antipsikotik atipikal) yang menenangkan sistem saraf sepatutnya mencukupi untuk memperbaiki tidur pada pesakit yang mudah teruja. Tetapi dalam kes AS, penggunaan ubat-ubatan tersebut penuh dengan kejadian kesan negatif. Oleh itu, doktor masih lebih suka pil tidur ringan, seperti Melatonin (ubat hormon semulajadi berdasarkan hormon tidur), yang diberikan kepada pesakit sejam sebelum tidur dalam jumlah 1 tablet, dan Diphenhydramine. Kekerapan pentadbiran dan dos yang ditentukan oleh doktor bergantung kepada keadaan dan umur pesakit.
Kadang-kadang pesakit dengan sindrom Angelman mempunyai masalah dengan penghadaman dan najis. Anda boleh memperbaiki najis anda dengan julap (sebaik-baiknya herba).
Atau anda boleh mendekati masalah secara berbeza, seperti yang dilakukan oleh doktor Amerika, berdasarkan beberapa kaedah merawat autisme, kerana banyak gejala ciri AS juga merupakan ciri autisme (impulsif, pergerakan sukarela, tindakan berulang, defisit perhatian, masalah komunikasi, dll.). Telah diperhatikan bahawa pengenalan hormon secretin, yang menormalkan pencernaan dan najis, mempunyai kesan positif terhadap perhatian pesakit, dan oxytocin membantu meningkatkan kebolehan kognitif dan ingatan kanak-kanak, dan tingkah laku yang betul.
Benar, hormon sahaja tidak mencukupi, terutamanya apabila ia berkaitan dengan kanak-kanak. Dalam sindrom Angelman, terapi tingkah laku, bekerja dengan ahli psikologi dan ahli terapi pertuturan (mengajar kaedah komunikasi bukan lisan dan bahasa isyarat) ditunjukkan. Pendidikan kanak-kanak tersebut hendaklah berdasarkan program individu dengan penyertaan guru terlatih khas, ahli psikologi dan ibu bapa. Malangnya, ini tidak boleh dilakukan di mana-mana, dan keluarga ditinggalkan bersendirian dengan masalah mereka.
Oleh kerana ramai pesakit muda dengan AS mengalami masalah otot dan sendi yang rendah, banyak perhatian diberikan kepada fisioterapi. Selalunya, doktor menggunakan aplikasi parafin, elektroforesis, dan terapi magnet.
Urut tonik aktif dan latihan khas latihan fizikal terapeutik akan membantu kanak-kanak yang sakit untuk berdiri di atas kakinya dan berjalan dengan yakin selepas beberapa ketika. Aquagymnastics amat berguna dalam hal ini, yang disyorkan untuk SA dalam air sejuk. Ia meningkatkan nada otot dan mengajar kanak-kanak untuk mengawal badannya dan menyelaraskan pergerakan.
Rawatan anticonvulsant
Simptom sindrom Angelman yang paling berbahaya ialah sawan yang serupa dengan epilepsi. Gejala ini diperhatikan dalam 80% pesakit, yang bermaksud bahawa kesemua mereka perlu diberi rawatan antikonvulsan yang berkesan.
Rawatan sawan epilepsi dijalankan dengan bantuan vitamin dan anticonvulsants. Dalam sindrom Angelman, disertai dengan sindrom sawan, vitamin kumpulan B, serta vitamin C, D dan E akan berguna. Tetapi menetapkan terapi vitamin sendiri dalam kes ini adalah sangat berbahaya, kerana pengambilan vitamin yang tidak terkawal boleh mengurangkan keberkesanan ubat antiepileptik dan mencetuskan sawan baru, lebih teruk dan berpanjangan.
Pemilihan ubat antikonvulsan dan preskripsi dos berkesannya juga perlu dilakukan oleh doktor pakar. Dia juga memutuskan sama ada satu ubat akan mencukupi atau sama ada pesakit perlu mengambil 2 atau lebih ubat untuk masa yang lama.
Bagi kebanyakan pesakit, doktor menetapkan ubat asid valproik (asid Valproik, Depakine, Convulex, Valparin, dll.), yang menghalang sawan dan meningkatkan mood dan keadaan mental pesakit.
Asid valproik boleh didapati dalam bentuk tablet, sirap dan larutan suntikan. Ubat yang paling popular ialah ubat pelepasan berpanjangan "Depakine" dalam tablet dan sebagai penyelesaian untuk pentadbiran intravena. Dos ubat ditentukan oleh doktor secara individu bergantung pada berat, umur dan keadaan pesakit.
Ubat ini diambil semasa makan 2 hingga 3 kali sehari. Purata dos harian ialah 20-30 mg setiap 1 kilogram berat badan pesakit, maksimum ialah 50 mg/kg sehari.
Kontraindikasi untuk digunakan. Jangan gunakan dalam kes disfungsi hati dan pankreas, diatesis hemoragik, hepatitis, porfiria dan hipersensitiviti kepada ubat.
Kesan sampingan termasuk gegaran tangan, gangguan pencernaan dan najis, dan perubahan berat badan.
"Topiramate" juga merupakan ubat pilihan untuk SA. Ia dihasilkan dalam bentuk tablet dan digunakan sebagai sebahagian daripada monoterapi dan dalam kombinasi dengan ubat lain.
Kaedah pentadbiran dan dos. Ambil tablet secara lisan tanpa mengira pengambilan makanan. Dos harian awal untuk orang dewasa ialah 25-50 mg, untuk kanak-kanak - 0.5-1 mg/kg. Setiap minggu, dos dinaikkan mengikut arahan doktor.
Ubat tidak boleh diambil semasa kehamilan dan penyusuan, serta dalam kes hipersensitiviti kepada komponennya. Dadah mempunyai banyak kesan sampingan yang berbeza.
Dadah yang mungkin ditetapkan oleh doktor untuk sindrom Angelman: Clomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, dll.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Perubatan tradisional dan homeopati
Perubatan tradisional, seperti persediaan homeopati, sudah tentu agak selamat, tetapi keberkesanan rawatan sedemikian untuk sindrom Angelman boleh dianggap kontroversi.
Walaupun rawatan rakyat masih boleh membantu dalam beberapa perkara. Kami bercakap tentang menghentikan sawan epilepsi. Dalam hal ini, rawatan herba boleh menjadi agak berkesan.
Kesan yang baik disediakan oleh koleksi ubat berdasarkan peony, licorice dan duckweed (komponen diambil dalam kuantiti yang sama). Herba perlu dikisar menjadi tepung. Selepas 2 minggu dari permulaan mengambilnya, anda boleh melihat penurunan ketara dalam kekerapan sawan.
Rebusan lavender (1 sudu teh setiap segelas air mendidih) juga berguna untuk kekejangan. Campuran direbus selama 5 minit dan diselitkan selama setengah jam. Ubat ini diambil pada waktu malam selama 14 hari.
Penyerapan berair (atau alkohol) motherwort dianggap berkesan untuk sawan epilepsi.
Daripada persediaan homeopati untuk mencegah sawan dalam sindrom Angelman, anda boleh menggunakan ubat berdasarkan chamomile dan motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, album Arsenicum. Tetapi perlu diambil kira bahawa hanya doktor homeopati boleh menetapkan dos ubat yang berkesan dan selamat dalam setiap kes tertentu.
Pencegahan
Seperti yang mungkin telah difahami oleh pembaca, ubat masih belum dapat mencegah mutasi gen dan keabnormalan kromosom lain, walau bagaimanapun, serta membetulkan keadaan. Ini boleh berlaku kepada sesiapa sahaja, kerana kanak-kanak dengan sindrom Angelman dilahirkan oleh ibu bapa yang sihat, dan genetik, yang kini merupakan salah satu cabang perubatan yang paling kurang dikaji, belum dapat menjelaskan perkara ini.
Satu-satunya perkara yang boleh dilakukan ialah mengambil pendekatan yang bertanggungjawab terhadap perancangan kehamilan, mendaftar dan menjalani peperiksaan tepat pada masanya. Tetapi sekali lagi, langkah sedemikian akan lebih mendidik daripada pencegahan, seperti mana-mana peperiksaan. Tetapi ibu bapa muda akan mengetahui terlebih dahulu apa yang perlu disediakan, dan sekiranya jawapan positif, mereka akan memutuskan sama ada mereka boleh mengambil tanggungjawab seperti membesarkan anak yang sakit.
Ramalan
Prognosis untuk sindrom Angelman bergantung pada sifat keabnormalan kromosom dan ketepatan masa pengesanannya. Yang paling teruk terjejas ialah kanak-kanak yang kromosom 15 mengandungi "jurang" dalam gen (penghapusan). Kemungkinan pesakit sedemikian berjalan dan bercakap sangat rendah. Kes-kes lain boleh diperbetulkan dengan pendekatan yang teliti dan kasih sayang untuk anak anda.
Malangnya, pesakit sedemikian tidak akan dapat menjadi ahli masyarakat sepenuhnya, walaupun pada hakikatnya mereka jauh dari bodoh, mereka memahami ucapan dan maknanya. Walau bagaimanapun, mereka akan menghadapi masalah dengan komunikasi sepanjang hayat mereka. Pesakit boleh diajar bahasa isyarat sejak kecil, tetapi mereka tidak boleh dipaksa untuk berkomunikasi menggunakan perkataan. Perbendaharaan kata pesakit "bercakap" terhad kepada minimum perkataan yang digunakan dalam kehidupan seharian (5-15 perkataan).
Bagi jangka hayat dan kesihatan umum pesakit dengan sindrom Angelman, angka di sini berubah-ubah di sekitar nilai purata. Pada masa dewasa, pesakit kebanyakannya menghadapi masalah kesihatan seperti scoliosis dan obesiti, yang, dengan pendekatan rawatan yang betul, tidak mengancam nyawa.