
Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Sindrom aper
Pakar perubatan artikel itu

Ulasan terakhir: 04.07.2025

 [
[ Punca Sindrom aper
Perkembangan sindrom diprovokasi oleh gangguan dalam struktur gen yang terletak pada kromosom 10. Gen ini bertanggungjawab untuk proses pemisahan jari, dan sebagai tambahan, untuk penutupan jahitan tengkorak tepat pada masanya.
Di samping itu, penyakit berjangkit yang dialami semasa kehamilan (seperti rubella atau tuberkulosis, sifilis atau selesema, serta meningitis), dan pendedahan ibu kepada sinar-X dianggap sebagai punca patologi. Selalunya, sindrom sedemikian dikesan pada kanak-kanak yang dilahirkan oleh ibu bapa yang lebih tua.
Patogenesis
Sindrom Apert adalah keturunan. Jenisnya adalah dominan autosomal (iaitu, dalam keluarga di mana salah seorang ibu bapa masa depan sakit, kebarangkalian mempunyai bayi dengan penyakit ini adalah 50-100%).
Mutasi unik dalam reseptor faktor pertumbuhan fibroblast 2 (FGFR2) mengakibatkan peningkatan bilangan sel progenitor yang berkembang di sepanjang laluan osteogenik. Ini akhirnya mengakibatkan peningkatan pembentukan matriks tulang subperiosteal dan pengerasan pramatang jahitan kranial semasa perkembangan janin. Susunan dan kadar pencairan jahitan menentukan tahap kecacatan dan ketidakupayaan. Apabila bahan jahitan telah sembuh, pertumbuhan tisu lain yang berserenjang dengan jahitan ini adalah terhad, dan tulang bercantum bertindak sebagai perancah tulang tunggal.
Bukti genetik pertama bahawa syndactyly dalam sindrom Apert adalah disebabkan oleh kecacatan pada reseptor faktor pertumbuhan keratinocyte (KGFR) adalah pemerhatian korelasi antara ekspresi KGFR dalam fibroblas dan keterukan syndactyly.
Amblyopia dan strabismus lebih biasa pada pesakit dengan mutasi FGFR2 Ser252Trp, dan atrofi cakera optik lebih biasa pada pesakit dengan mutasi FGFR2 Pro253Arg. Pesakit dengan mutasi FGR2 Ser252Trp mempunyai prevalens kecacatan penglihatan yang jauh lebih tinggi berbanding pesakit dengan mutasi FGFR2 Pro253Arg.
Gejala Sindrom aper
Beberapa manifestasi penyakit sudah jelas kelihatan pada kelahiran bayi, kerana ia berkembang di dalam rahim ibu. Antara simptom utama sindrom:
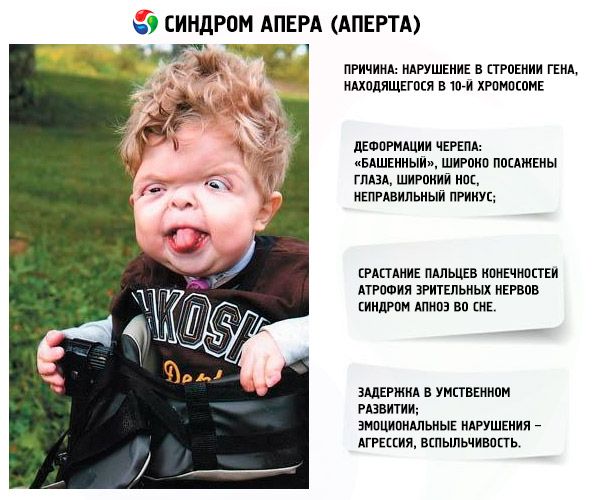
- Tengkorak itu cacat - ia membentang tinggi, memperoleh penampilan "menara"; di samping itu, mata ditetapkan lebar dan sedikit membonjol (kerana saiz soket mata berkurangan); hidung melebar dan gigitan yang tidak betul terbentuk (gigi atas menonjol secara berlebihan);
- Jari-jari anggota badan bercantum sepenuhnya (terutamanya jari manis, serta jari tengah dan jari telunjuk) dan kelihatan seperti selaput kulit atau gabungan tulang yang lengkap; di samping itu, jari tambahan mungkin tumbuh;
- Terencat akal (tidak berlaku pada semua orang);
- Atrofi saraf optik, akibatnya ketajaman penglihatan berkurangan (dalam beberapa kes, kehilangan penglihatan lengkap berkembang);
- Peningkatan tekanan intrakranial, yang berlaku akibat penutupan pramatang jahitan tengkorak, menunjukkan dirinya dalam bentuk sakit kepala dan muntah dengan loya;
- Oleh kerana rahang atas masih kurang berkembang, masalah pernafasan timbul;
- Sindrom apnea tidur adalah kejadian biasa.
- Manifestasi emosi - pencerobohan, kekurangan kekangan, mudah marah yang kuat.
Komplikasi dan akibatnya
Diagnostik Sindrom aper
Untuk membuat diagnosis, langkah-langkah berikut diperlukan:
- Doktor harus menganalisis aduan pesakit, serta sejarah perubatan. Adalah perlu untuk mengetahui sama ada terdapat kes patologi yang sama dalam keluarga;
- Pemeriksaan neurologi untuk menilai bentuk tengkorak dan perkembangan intelek pesakit (soal selidik khas serta temu bual);
- Pemeriksaan fundus untuk mengesan kehadiran gejala peningkatan tekanan intrakranial (bengkak cakera optik, serta kabur tepinya);
- Untuk menilai keadaan tengkorak, X-ray dilakukan;
- Pengimejan resonans komputer dan magnetik kepala untuk memeriksa struktur otak dan tengkorak lapisan demi lapisan, untuk menentukan kehadiran gejala gabungan pramatang jahitan kranial, dan di samping itu, hidrosefalus (disebabkan oleh peningkatan tekanan intrakranial, cecair serebrospinal yang berlebihan terkumpul (ini adalah cecair serebrospinal yang menggalakkan proses metabolik otak, serta));
- X-ray kaki dan tangan untuk menentukan punca gabungan jari (ini penting untuk merancang campur tangan pembedahan berikutnya);
- Perundingan dengan pakar genetik perubatan dan pakar bedah saraf mungkin ditetapkan.
Ujian
Analisis genetik mutasi biasa yang berlaku dalam gen jenis FGFR2 sedang dijalankan.
Diagnosis pembezaan
Sindrom ini mesti dibezakan daripada patologi genetik lain di mana craniosynostosis diperhatikan. Ini adalah penyakit seperti sindrom Pfeiffer, Crouzon, Saethre-Chotzen dan Carpenter. Kaedah ujian genetik molekul digunakan untuk mengecualikan anomali ini.
Siapa yang hendak dihubungi?
Rawatan Sindrom aper
Pembedahan dianggap satu-satunya rawatan yang berkesan untuk sindrom Apert - ia membantu membetulkan kecacatan fizikal individu dan juga membetulkan terencat akal.
Semasa prosedur ini, jahitan koronal ditutup untuk mengelakkan kemungkinan kecederaan otak. Teknik yang paling biasa ialah gangguan kraniofasial, yang melibatkan tarikan tengkorak berperingkat. Pembedahan ortodontik dan/atau ortognatik dilakukan untuk membuang kecacatan muka individu.
Di samping itu, pesakit menjalani pembetulan pembedahan gabungan jari.
Rujukan
Genetik Perubatan. Panduan Negara. Disunting oleh Ginter EK, Puzyrev VP GEOTAR-Media. 2022