
Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Sindrom Treacher Collins
Pakar perubatan artikel itu

Ulasan terakhir: 04.07.2025

Gangguan intrauterin dalam proses perkembangan tulang menyebabkan kecacatan kraniofasial yang serius, dan salah satu daripada jenis patologi tersebut ialah sindrom Treacher Collins (TCS) atau mandibulofasial, iaitu dysostosis maxillofacial.
Kod penyakit mengikut ICD 10: kelas XVII (anomali kongenital, ubah bentuk dan gangguan kromosom), Q75.4 - dysostosis mandibulofacial.
Punca Sindrom Treacher Collins
Sindrom ini dinamakan sempena pakar oftalmologi British yang cemerlang Edward Treacher Collins, yang menerangkan ciri-ciri utama patologi lebih daripada seratus tahun yang lalu. Walau bagaimanapun, doktor Eropah lebih kerap memanggil jenis anomali tulang muka dan rahang ini sebagai penyakit atau sindrom Franceschetti - berdasarkan penyelidikan meluas pakar mata Switzerland Adolf Franceschetti, yang memperkenalkan istilah "mandibulofascial dysostosis" pada pertengahan abad yang lalu. Dalam kalangan perubatan, nama sindrom Franceschetti-Collins juga digunakan.
Sindrom Treacher Collins disebabkan oleh mutasi dalam gen TCOF1 (di lokus kromosom 5q31.3-33.3), yang mengkodkan fosfoprotein nukleolar yang bertanggungjawab untuk pembentukan bahagian kraniofasial embrio manusia. Akibat penurunan pramatang dalam jumlah protein ini, biogenesis dan fungsi rRNA terganggu. Menurut ahli genetik dari program penyelidikan Genom Manusia, proses ini membawa kepada pengurangan percambahan sel embrio puncak neural - rabung di sepanjang alur saraf, yang menutup ke dalam tiub saraf semasa perkembangan embrio.
Pembentukan tisu muka berlaku disebabkan oleh transformasi dan pembezaan sel bahagian atas (kepala) puncak saraf, yang berhijrah di sepanjang tiub saraf ke kawasan gerbang cawangan pertama dan kedua embrio. Dan kekurangan sel-sel ini menyebabkan ubah bentuk kraniofasial. Tempoh kritikal untuk berlakunya anomali adalah dari 18 hingga 28 hari selepas persenyawaan. Setelah selesai penghijrahan sel-sel puncak saraf (pada minggu keempat kehamilan), hampir semua tisu mesenchymal longgar di kawasan muka terbentuk, yang kemudiannya (dari 5 hingga 8 minggu) membezakan menjadi tisu rangka dan penghubung semua bahagian muka, leher, laring, telinga (termasuk telinga dalam) dan masa depan.
Patogenesis
Patogenesis sindrom Treacher Collins selalunya bersifat keluarga, dan anomali itu diwarisi secara dominan autosomal, walaupun terdapat kes transmisi resesif autosomal kecacatan (dengan mutasi dalam gen lain, khususnya, POLR1C dan POLR1D). Perkara yang paling tidak dapat diramalkan mengenai dysostosis maxillofacial ialah mutasi itu diwarisi oleh kanak-kanak hanya dalam 40-48% kes. Iaitu, dalam 52-60% pesakit, penyebab sindrom Treacher Collins tidak dikaitkan dengan kehadiran anomali dalam keluarga, dan dipercayai bahawa patologi berlaku akibat mutasi gen sporadis de novo. Kemungkinan besar, mutasi baru adalah akibat kesan teratogenik pada janin semasa kehamilan.
Antara punca teratogenik sindrom ini, pakar menamakan dos besar etanol (etil alkohol), radiasi, asap rokok, sitomegavirus dan toxoplasma, serta racun herba berasaskan glifosat (Roundal, Glyfor, Tornado, dll.). Dan senarai faktor iatrogenik termasuk ubat jerawat dan seborrhea dengan asid 13-cis-retinoik (Isotretinoin, Accutane); ubat anticonvulsant Phenytoin (Dilantin, Epanutin); ubat psikotropik Diazepam, Valium, Relanium, Seduxen.
Gejala Sindrom Treacher Collins
Untuk sebahagian besar, tanda-tanda klinikal dysostosis mandibulofascial dan tahap ekspresinya bergantung pada ciri-ciri manifestasi mutasi gen. Dan tanda-tanda pertama anomali ini dalam kebanyakan kes dapat dilihat pada kanak-kanak sejurus selepas kelahiran: wajah dengan sindrom Treacher Collins mempunyai penampilan yang khas. Selain itu, anomali morfologi biasanya dua hala dan simetri.
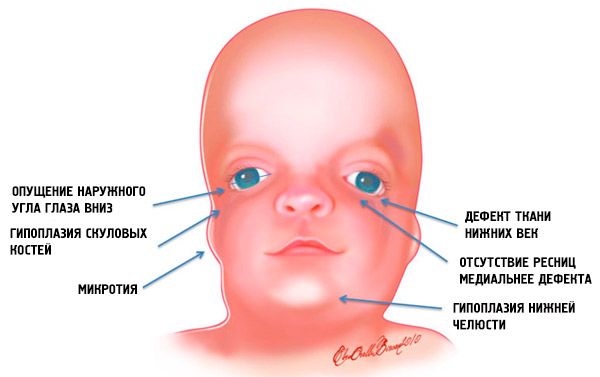
Gejala yang paling jelas bagi sindrom Treacher Collins ialah:
- kurang pembangunan (hipoplasia) tulang muka tengkorak: zygomatic, proses zygomatic tulang depan, plat pterygoid sisi, sinus paranasal, rahang bawah dan tonjolan epifisis tulang (condyles);
- kurang perkembangan tulang rahang bawah (micrognathia) dan sudut rahang bawah yang lebih tumpul daripada biasa;
- hidung bersaiz normal, tetapi kelihatan besar disebabkan oleh hipoplasia gerbang superciliary dan keterbelakangan atau ketiadaan gerbang zigomatik di kawasan temporal;
- celah mata adalah ke bawah, iaitu, bentuk mata tidak normal, dengan sudut luar terkulai ke bawah;
- kecacatan kelopak mata bawah (coloboma) dan ketiadaan separa bulu mata pada mereka;
- aurikel berbentuk tidak teratur dengan pelbagai penyimpangan, termasuk lokasinya di sudut rahang bawah, ketiadaan cuping, fistula buta antara tragus telinga dan sudut mulut, dsb.;
- penyempitan atau penutupan (atresia) saluran pendengaran luaran dan anomali ossikel telinga tengah;
- ketiadaan atau hipoplasia kelenjar air liur parotid;
- hipoplasia pharyngeal (penyempitan pharynx dan saluran pernafasan);
- tidak bercantum lelangit keras (lelangit sumbing), serta ketiadaan, pemendekan atau imobilitas lelangit lembut.
Anomali anatomi sedemikian dalam semua kes mempunyai komplikasi. Ini adalah gangguan pendengaran berfungsi dalam bentuk kehilangan pendengaran konduktif atau pekak lengkap; kecacatan penglihatan akibat pembentukan bola mata yang tidak betul; kecacatan lelangit menyebabkan kesukaran untuk memberi makan dan menelan. Terdapat gangguan oklusi gigi (maloklusi) yang dikaitkan dengan kecacatan rahang, yang seterusnya, menyebabkan masalah dengan mengunyah dan artikulasi. Patologi lelangit lembut menjelaskan suara hidung.
Komplikasi dan akibatnya
Akibat anomali maxillofacial dalam sindrom Treacher Collins ialah semasa kelahiran kebolehan intelek kanak-kanak adalah normal, tetapi disebabkan kecacatan pendengaran dan gangguan lain, terencat akal sekunder diperhatikan.
Di samping itu, kanak-kanak yang mengalami kecacatan sedemikian sangat merasakan rendah diri dan menderita, yang memberi kesan negatif kepada sistem saraf dan jiwa mereka.
Diagnostik Sindrom Treacher Collins
Diagnosis sindrom Treacher Collins selepas bersalin pada asasnya berdasarkan tanda-tanda klinikal. Dysostosis kraniofasial mudah dikenal pasti apabila sindrom ekspresif sepenuhnya, tetapi apabila gejala patologi yang dinyatakan secara minimum, masalah dengan menubuhkan diagnosis yang betul mungkin timbul.
Dalam kes ini, perhatian khusus harus diberikan kepada penilaian semua fungsi yang berkaitan dengan anomali, terutamanya yang menjejaskan pernafasan (disebabkan oleh risiko apnea tidur). Keberkesanan penyusuan dan ketepuan oksigen hemoglobin juga harus dinilai dan dipantau.
Kemudian, pada hari ke-5-6 selepas kelahiran, tahap kerosakan pendengaran perlu ditentukan menggunakan ujian audiologi, yang harus dijalankan di hospital bersalin.
Pemeriksaan ditetapkan, di mana diagnostik instrumental dilakukan oleh fluoroskopi dismorfologi kraniofasial; pantomografi (panoramik X-ray struktur tulang tengkorak muka); tomografi terkira tengkorak penuh dalam pelbagai unjuran; CT atau MRI otak untuk menentukan keadaan saluran pendengaran dalaman.
Diagnosis terawal - pranatal - anomali maxillofacial dengan kehadiran sindrom Treacher Collins dalam sejarah keluarga adalah mungkin dengan biopsi vilus korionik pada 10-11 minggu kehamilan (prosedur mengancam keguguran dan jangkitan rahim).
Ujian darah juga diambil daripada ahli keluarga; pada 16-17 minggu kehamilan, cecair amniotik dianalisis (amniosentesis transabdominal); pada 18-20 minggu kehamilan, fetoskopi dilakukan dan darah diambil dari saluran janin plasenta.
Tetapi selalunya, ultrasound digunakan dalam diagnosis pranatal sindrom ini pada janin (pada 20-24 minggu kehamilan).
Ujian apa yang diperlukan?
Diagnosis pembezaan
Kaedah yang sama digunakan oleh pakar apabila diagnostik pembezaan diperlukan untuk mengenali sindrom Treacher Collins yang ringan dan membezakannya daripada anomali kongenital tulang kraniofasial yang lain, khususnya: Apert, Crouzon, Nager, Peters-Hewels, sindrom Hellermann-Steph, serta sindrom hemifacial premature of the craniofacial, hypertelordenharism, hypertelordenharism. (craniosynostosis) atau gangguan gabungan tulang muka (craniosynostosis).
Siapa yang hendak dihubungi?
Rawatan Sindrom Treacher Collins
Seperti dalam semua kes kecacatan kongenital yang ditentukan secara genetik, rawatan bentuk sindrom Treacher Collins yang teruk adalah paliatif secara eksklusif, kerana tiada kaedah terapeutik untuk patologi tersebut. Spektrum dan tahap ubah bentuk dalam sindrom ini adalah luas dan, oleh itu, sifat dan intensiti campur tangan perubatan juga mempunyai banyak pilihan.
Alat bantu pendengaran digunakan untuk membetulkan dan memperbaiki pendengaran, dan sesi terapi pertuturan digunakan untuk memperbaiki pertuturan.
Campur tangan pembedahan diperlukan pada usia awal dalam kes penyempitan saluran pernafasan yang teruk (trakeostomi dilakukan) dan laring (gastrostomi dilakukan untuk memberi makan). Pembetulan pembedahan lelangit juga mungkin diperlukan.
Pembedahan memanjangkan mandibular dilakukan pada usia 2-3 tahun atau lebih baru. Pembinaan semula tisu lembut termasuk pembetulan koloboma kelopak mata bawah dan pembedahan plastik aurikular.
Pencegahan
Ramalan
Apakah prognosis untuk patologi ini? Ia bergantung kepada tahap ubah bentuk dan keamatan gejala. Sindrom Treacher Collins adalah diagnosis sepanjang hayat.
 [ 25 ]
[ 25 ]